Please see Instructions for Use.


For Personal Use

For Healthcare Professionals

For Schools

For Businesses

SWAB.

SWIRL.
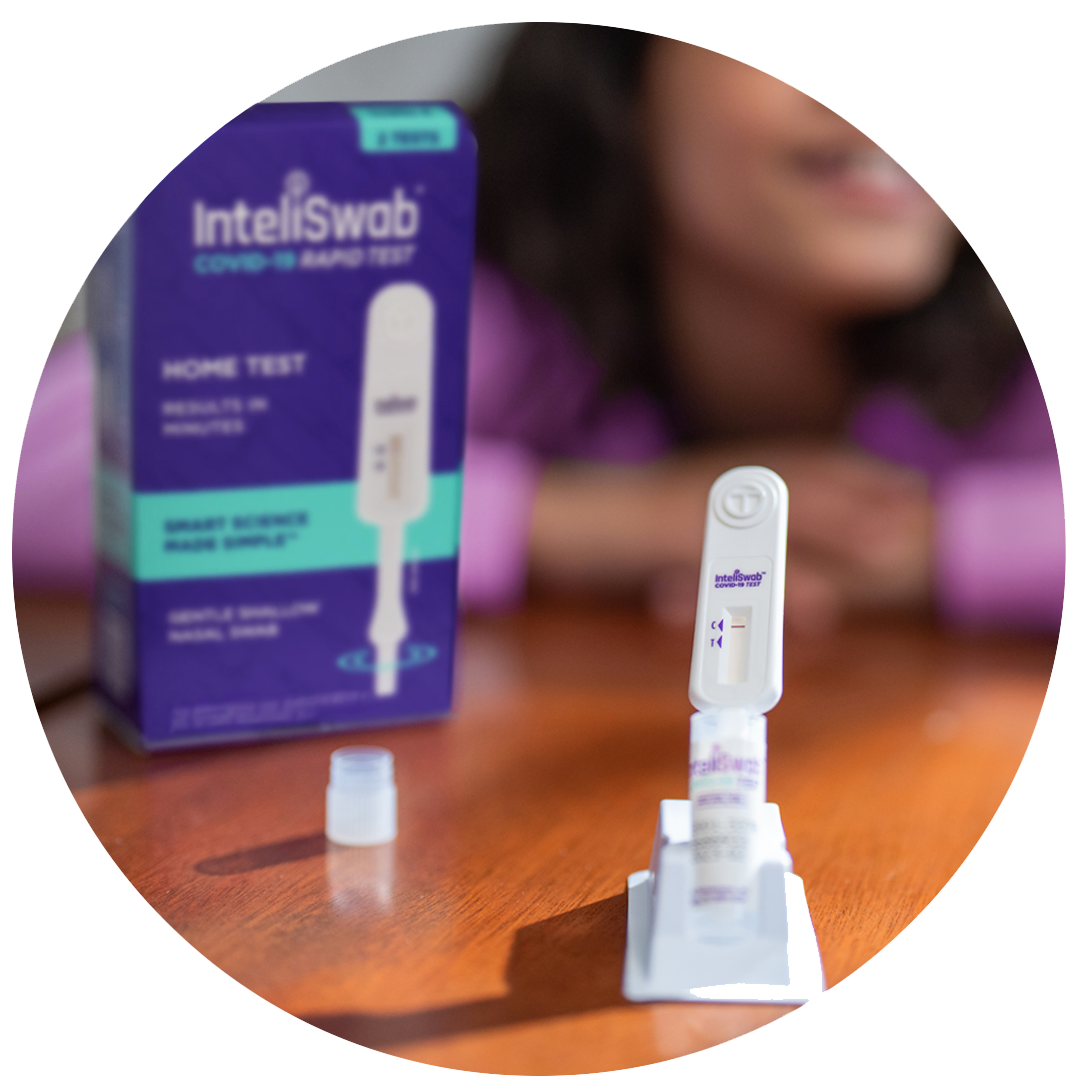
SEE.



Former Pennsylvania Health Secretary Rachel Levine refers to [InteliSwab®] tests as “a game changer” in the fight against COVID-19.

“[OraSure Technologies’] efforts were praised by President Joe Biden during a stop in the Lehigh Valley last summer.”

For over 30 years, the innovators at OraSure have worked with the FDA to deliver critical diagnostic tests that protect public health.

InteliSwab® is just one of two COVID-19 tests selected by the U.S. government to supply schools nationwide.
